🧫 Sequencing a peculiar K. pneumoniae isolate
ARGs found in a weird looking KP culture
Antibiotic resistance is a growing challenge, and sometimes an interesting case lands on your bench thanks to great collaborations. That was exactly the situation with a particularly stubborn Klebsiella pneumoniae that our colleagues at the National Institute for Research and Development in Microbiology and Immunology “Cantacuzino” had cultivated. After observing its unusual growth characteristics and alarmingly high antibiotic resistance, they kindly forwarded it to us for a deeper dive using whole-genome sequencing. My aim was to pinpoint the specific antibiotic resistance genes (ARGs) this isolate was using for its defense and to understand their full genomic context – especially whether these genes were on plasmids, which can often be tricky to fully characterize.
💧 Wetlab phase
- 1. DNA Extraction
- Bacterial DNA was extracted directly from the bacterial culture using QIAGEN’s QiAMP Microbiome Kit (manual extraction, bead beating as cell lysis method), yielding approximately 80 ng/µL.
- 2. Library prep
- SQK-LSK110 ligation sequencing kit, NEBNext Ultra II End repair module, NEBNext Quick Ligation Module and AMPure XP Beads were used for library preparation.
- 3. Sequencing
- Loading the library on an R9.4.1 flowcell on the MinION Mk1B was straight forward. 13.6 fmoles of library used. The flowcell used was being stored at 4°C for months after its previous run, but it still had a decent amount of active pores, at least on the initial check - 920 out of the maximum advertised amount of 2048.
- 4. Basecalling
- The next day, after 19 hours of run time, we were left with approx. 580 pores and enough reads (612k passed) to stop the run. The real-time basecalling (HAC model) generated 4Gb, with an approx. N50 of 16Kb. Estimated coverage of ~742x.
🧬 Data analysis
I wanted to test RRWick’s new tool - Trycycler. It’s designed to generate high-quality bacterial consensus assemblies from long-read sequencing data. Unlike traditional assemblers that rely on a single method, Trycycler takes a hybrid approach by combining multiple assemblies of the same dataset, leveraging their strengths to produce a more accurate and contiguous final assembly. It seemed like the perfect moment to test this method so I had to give it a go.
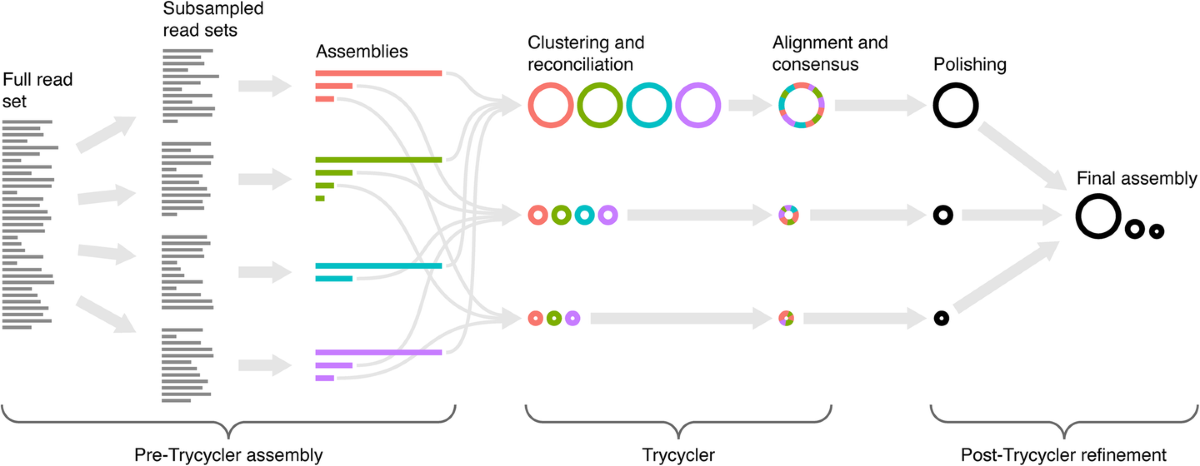 Trycycler workflow. Credits: Trycycler Github
Trycycler workflow. Credits: Trycycler Github
Running EPI2ME’s WIMP
Just to make sure our reads are K. pneumoniae.

QC and filtering using Filtlong
QC scores were good, we kept fragments longer than 1Kb to help with the assembly.1 2 3
zcat *.fastq.gz > KP9_sup_seq.fastq fastqc KP9_sup_seq.fastq filtlong –min_length 1000 –keep_percent 95 KP9_sup_seq.fastq > KP9_sup_filtrat.fastq
Subsampling reads
This step creates the assemblies.We used Flye, Miniasm/Minipolish and Raven.1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
trycycler subsample --reads KP9_sup_filtrat.fastq --out_dir subsets --genome_size 5.5m --threads 48 #Flye: for s in 01 04 07 10; do flye --nano-hq subsets/sample_”$s”.fastq --threads 52 --out_dir assemblies/assembly”$s” done #Miniasm/Minipolish: for s in 02 05 08 11; do minimap2 -x ava-ont -t 52 subsets/sample_”$s”.fastq subsets/sample_”$s”.fastq > overlaps.paf miniasm -f subsets/sample_”$s”.fastq overlaps.paf > assembly.gfa minipolish -t 52 subsets/sample_”$s”.fastq assembly.gfa > polished.gfa any2fasta polished.gfa > assemblies/assembly”$s”/assembly”$s”.fasta done #Raven: for s in 03 06 09 12; do raven --threads 52 --disable-checkpoints subsets/sample_”$s”.fastq > assemblies/assembly”$s”/assembly”$s”.fasta done
Manual curation and clustering
I used Bandage to check the contig circularity. It looked fine, with a clear circular chromosome and what appeared to be 2 plasmids.
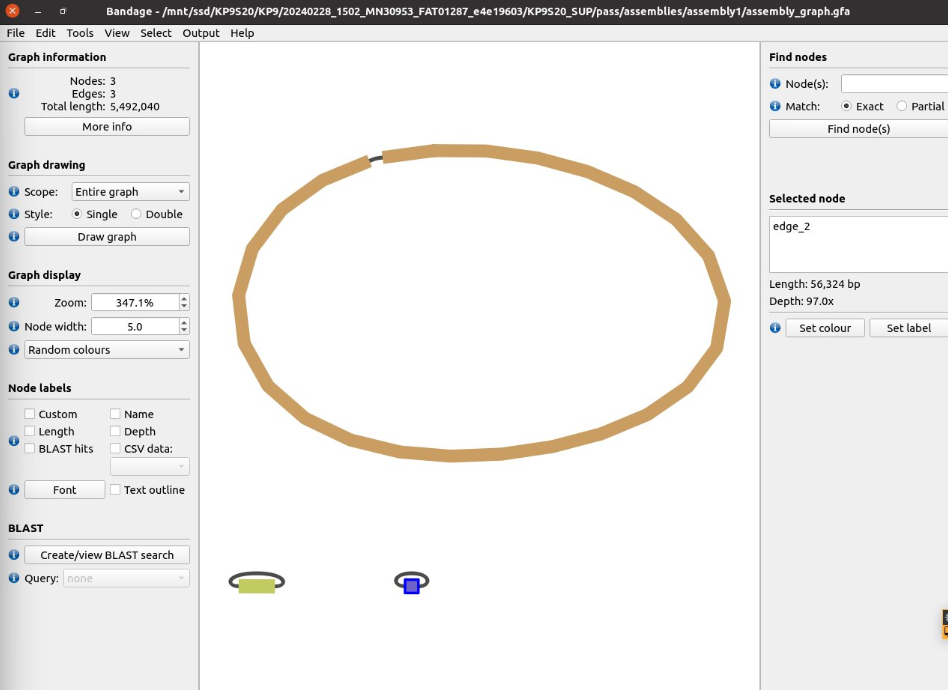
Clustering the assemblies was straight-forward using Trycycler.1
trycycler cluster --threads 48 --assemblies assemblies/*.fasta --reads KP9_sup_filtrat.fastq --out_dir cluster
I then looked at FigTree to identify out of place contigs. The clustering was succesful - 4 clearly defined clusters - a chromosome of ~5.3Mb and 3 smaller contigs of ~153Kb, ~56Kb and ~4Kb respectively, which were probably plasmids. I ended up removing the D contig in the 3rd cluster since its size was very different to the other ones.

- Alignment and consensus generation
Before generating the consensus sequence, the contigs have to be reconciled, aligned and partitioned. Reconciling contigs refers to the process of merging or resolving discrepancies between overlapping contigs, whereas the multiple sequence alignment (MSA) aligns the fragments against each other to identify regions of similarity/difference. Partinioning organizes the sequences into distinct regions which can be processed independently, improving accuracy. Contig reconciliation was the only step that needed manual processing, having me remove a few contigs that had low pairwise identities to one another. I only kept those with higher than 99% pairwise identity. The latter steps were as straightforward as they can get.1 2 3 4 5
#Ran these separately for all the clusters trycycler reconcile --threads 48 --reads KP9_sup_filtrat.fastq --cluster_dir clusters/cluster_00* trycycler msa –threads 48 –cluster_dir clusters/cluster_00* trycycler partition –threads 48 –reads KP9_sup_filtrat.fastq –cluster_dir clusters/cluster_00* trycycler consensus –threads 48 –cluster_dir clusters/cluster_00*
- Long and short read polishing
Using some short Illumina reads from our friends over at the Cantacuzino Institute I was able to take the clusters not only through Medaka for long-read polishing, but also Polypolish.1 2 3 4 5 6 7 8 9 10 11 12 13
#Medaka for all the clusters, renaming the files and cleaning the outputs for c in clusters/cluster_*; do medaka_consensus -i “$c”/4_reads.fastq -d “$c”/7_final_consensus.fasta -o “$c”/medaka -m r941_min_sup_g507 -t 48 mv “$c”/medaka/consensus.fasta “$c”/8_medaka.fasta rm -r “$c”/medaka “$c”/*.fai “$c”/*.mmi done #Combining the clusters into one single .FASTA cat clusters/cluster_*/8_medaka.fasta > KP9S20_consensus.fasta #Polypolish using shortreads bwa index KP9S20_consensus.fasta bwa mem -t 48 -a KP9S20_consensus.fasta shortreads/kp9_sr1.fastq.gz > shortreads/pp_align_1.sam bwa mem -t 48 -a KP9S20_consensus.fasta shortreads/kp9_sr2.fastq.gz > shortreads/pp_align_2.sam polypolish KP9S20_consensus.fasta shortreads/pp_align_1.sam shortreads/pp_align_2.sam > shortreads/polypolishkp9.fasta
🦠 ARG detection
To identify ARGs I’ve used another awesome tool - StarAMR which is able to scan the .FASTA against ResFinder/PointFinder/PlasmidFinder DBs, making a snazzy report of known antimicrobial resistance genes it detects.
1
staramr search polypolishkp9.fasta --output-dir ./AMR
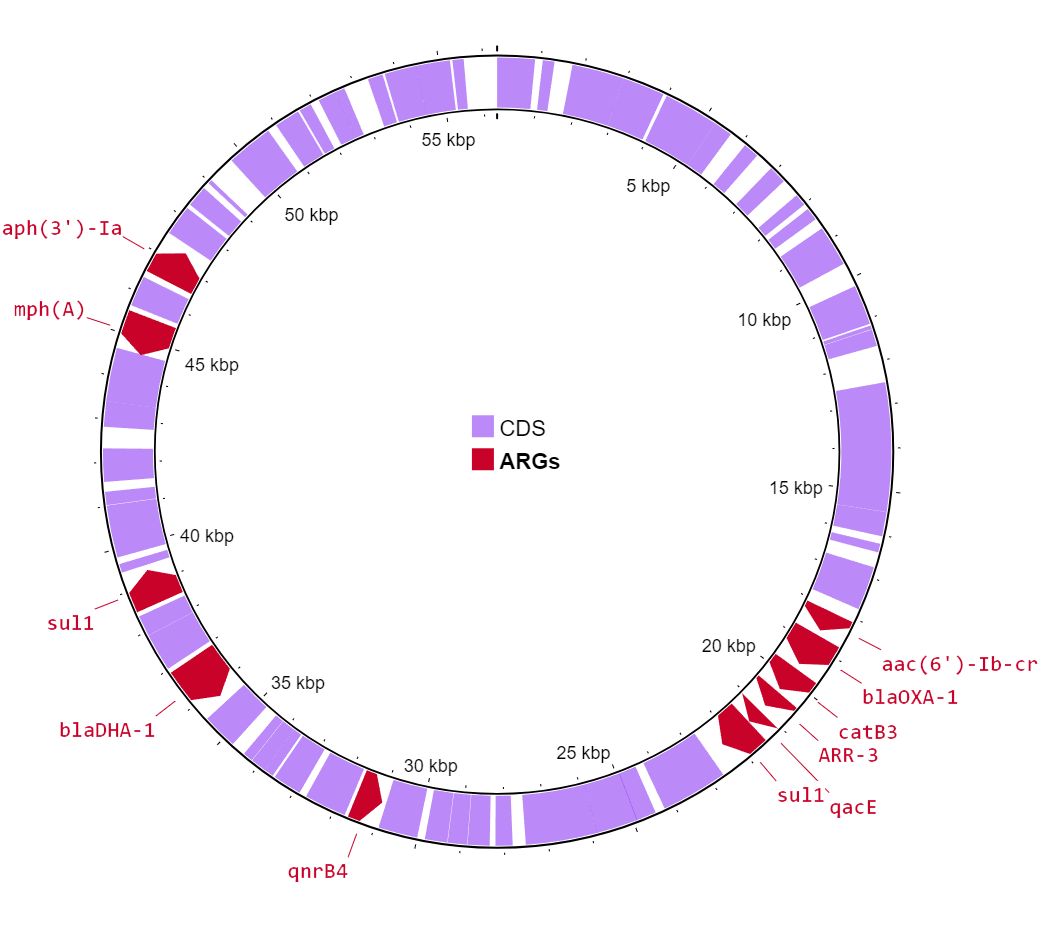
In the chromosome, StarAMR spotted blaTEM-1b, which is linked to resistance against piperacillin. The more notable ARGs were found in the plasmid - qnrB4, blaOXA-1, aac(6’)-Ib-cr and aph(3’)-Ia, which gives resistance to multiple antibiotics. The coolest (and a bit worrying) part? It found blaDHA-1, which hasn’t been documented in my region before.
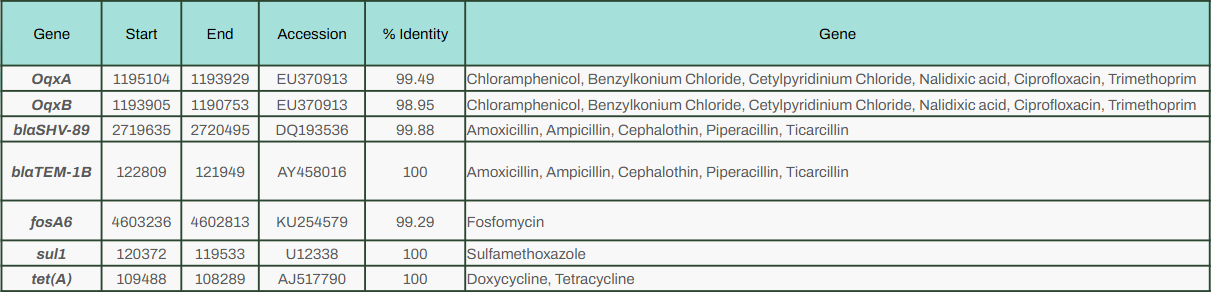 ARGs in the chromosome
ARGs in the chromosome 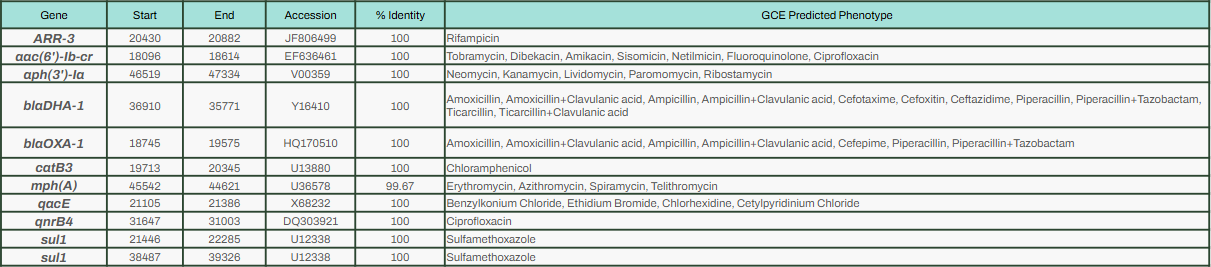 ARGs in the smaller, IncR plasmid
ARGs in the smaller, IncR plasmid
I also found this super cool and easy to use web interface that lets you visualise and annotate bacterial genomes, with interactive circule and linear genome maps - Proksee. I initially used pyCirclize to make some circos but this is just super fun to use. I’ll make a separate journal entry about it since the whole Python deal is interesting enough to have its own post.
Anyway, here are the circos generated with Proksee, as well as a zoomed view of the blaDHA-1 area of the plasmid, with some mobile genomic elements (MGEs) I had Proksee annotate for me.

📖 From a “weird bug” to a regional first
The work on this particular Klebsiella pneumoniae isolate, especially the successful identification of the blaDHA-1 gene on one of its plasmids – a first for this species in Romania – didn’t just end with the analysis.
We were pleased to be able to share these findings as a presentation at the 2024 AMLR Conference (The Romanian Association of Laboratory Medicine). Furthermore, this study detailing the genomic analysis and the blaDHA-1 discovery was also published in the Romanian Journal of Laboratory Medicine. It’s always good when detailed lab work and bioinformatics can contribute to the broader scientific record on important topics like antibiotic resistance.
You can read the abstract here, on page 77.
 AMLR 2024 Conference
AMLR 2024 Conference
🤔 Final thoughts
Trycyler worked great and was rather easy to use. The idea behind it is very solid too, improving accurancy over other tools like Unicycler. Learned a lot from this and found some cool ARGs. Time to dig deeper and try to understand where this Klebsiella pneumoniae isolate got its weird plasmid from.